
Introduction
The French neurologist Octave Crouzon initially identified Crouzon disease as one of the forms of craniofacial dysostosis in 1912. It is characterized by a triad of cranial deformities, facial anomalies, and exophthalmia. Complete penetrance and variable expressivity characterize the autosomal dominant inherited syndrome known as Crouzon syndrome.1 The disease is mainly brought on by damaged mesenchyme and ectoderm, which are brought on by chromosomal loci 10q25–10q26 mutations in the FGFR-2 gene. 50% of cases are caused by novel mutations and lack a family history, despite being hereditary. Premature fusion of the coronal and sagittal sutures, which starts in the first year of life, is the disease's hallmark. The potential for growth is limited once the seams close. Multiple suture synostoses, on the other hand, frequently cause the skull base to close too soon, which can lead to upper airway obstruction, maxillary hypoplasia, a shallow orbit, and midfacial hypoplasia. 2 Mandibular prognathism, upper tooth crowding, and a V-shaped dental arch in the maxilla are examples of intraoral manifestations. 3 Additionally visible are a uvula and a narrow, high, or cleft palate. There have been isolated reports of peg-shaped and widely spaced teeth, oligodontia, and macrodonia. 2, 3
Worldwide, one in every 25,000 births has the Crouzon syndrome.4 Roughly 4:8% of all cases of craniosynostosis are caused by Crouzon syndrome. 5 Neither race nor gender preference is known. Muenke, Carpenters, Apert, and Pfeiffer syndromes are included in the differential diagnosis of Crouzon syndrome. 6 Unless early closure of the cranial suture lines hinders brain development, mental retardation is not a defining characteristic. 7 This is a case report of a male patient, age 25, who has Crouzon syndrome.
Case Report
A 25-year-old male patient reported to the department of oral medicine with a chief complaint of forwardly placed lower jaw with respect to upper jaw. History revealed that the patient had difficulty in speech as well as had esthetic concerns. Medical history reveals that the patient had breathing difficulty and snoring from the age of 4 years and had undergone nasal surgery 8 years back.
On extra oral examination, the patient had maxillary hypoplasia (dish face deformity), hypertelorism and exophthalmos (Figure 1a). Patient also had turricephaly/oxycephaly/tower shaped head, saddle nose deformity, parrot beak (Figure 1b) and mandibular prognathism(Figure 1b).
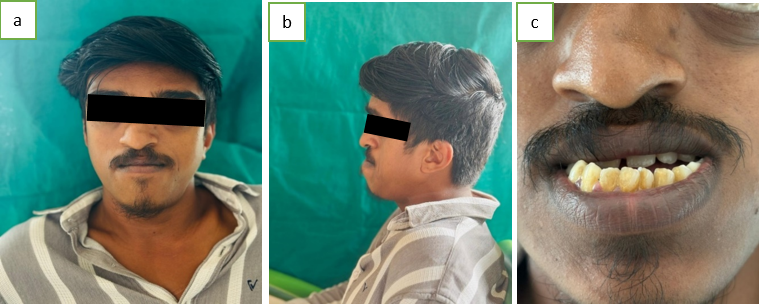
Intraoral examination revealed high arched palate, crowding of teeth, clinically missing 15,25 and anterior and posterior crossbite (Figure 1c).
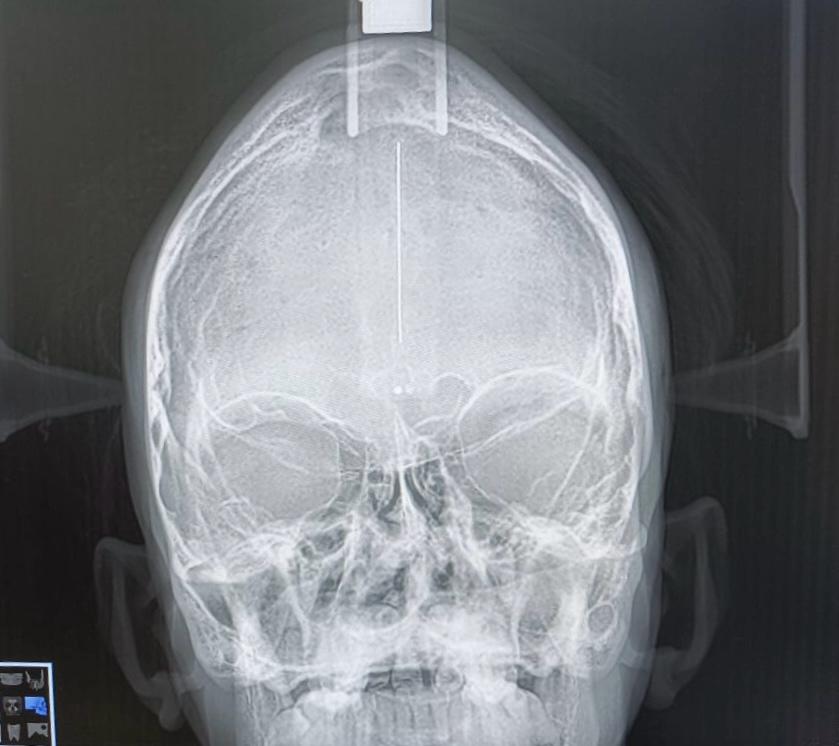
PA skull (Figure 2) shows craniofacial synostosis with copper beaten skull appearance indicating remodeling of brain due to increased intracranial pressure due to fused sutures.
Discussion
Crouzon syndrome is the second most common craniosynostosis syndrome and a relatively rare disease.8 Family history is a significant factor because of the autosomal inheritance pattern. The clinical diagnosis of Crouzon syndrome is aided by the worsening of facial deformities during the first two years of life. For the clinical diagnosis, the remainder of a newborn's medical history is less significant. An infant with Crouzon syndrome may present in a variety of ways, ranging from mild forms with midfacial weakness to severe forms with prominent midfacial and ocular abnormalities and multiple fused cranial sutures. These kids rarely have intellectual disabilities, and upper airway obstruction may be the source of their respiratory distress.9 If treatment is not received for increased intracranial pressure, optic atrophy can result in blindness.
Treatment of Crouzon syndrome requires a multidisciplinary approach and early diagnosis is important. Releasing cranial synostotic sutures is preferred during the first year of life to allow for adequate cranial volume and to support brain growth and expansion. To achieve the best possible results, skull reshaping may need to be repeated as the child grows.10 If necessary, midfacial advancement and jaw surgery can be performed to provide adequate orbital volume and reduce exophthalmoses to correct the occlusion to an appropriate functional position, thus providing a more normal appearance. In this case, the patient was diagnosed with Crouzon syndrome. Maxillary hypoplasia (cup face deformity), hypertelorism, and exophthalmos were noted on extraoral examination (Figure 1a). The patient also had turricephaly/oxycephaly/rook head, saddle nose deformity, parrot beak, and mandibular prognathism (Figure 1b). Intraoral examination revealed a high arched palate, crowding of teeth, clinically absent 15.25, and anterior and posterior crossbite (Figure 1c). The PA skull (Figure 2) shows craniofacial synostosis with copper-beaten skull appearance, indicating brain remodeling due to increased intracranial pressure caused by bonded sutures.
Conclusion
Because Crouzon syndrome causes facial expression impairment and other complications like breathing issues, aspiration pneumonia, vision problems, and mental retardation, it is important to treat it as soon as possible. These patients have the potential to be useful and contributing members of society given the right care. Comprehending these irregularities is imperative for the dental team to ensure that the patient receives the best care possible and to make the appropriate referrals.